Summary of service
The identification of orthologs is an important cornerstone for many comparative, evolutionary and functional genomics analyses. Yet, the true evolutionary history of genes is generally unknown. Because of the wide range of possible applications and taxonomic interests, benchmarking of orthology predictions remains a difficult challenge for methods developers and users.
This community developed web-service aims at simplifying and standardizing orthology benchmarking. And for the users, the benchmarks provide a way to identify the most effective methods for the problem at hand.
This website is free and open to all users and there is no login requirement.
The associated paper to the service has been published open access in Nature Methods. If you use the orthology benchmark service, please consider citing it.
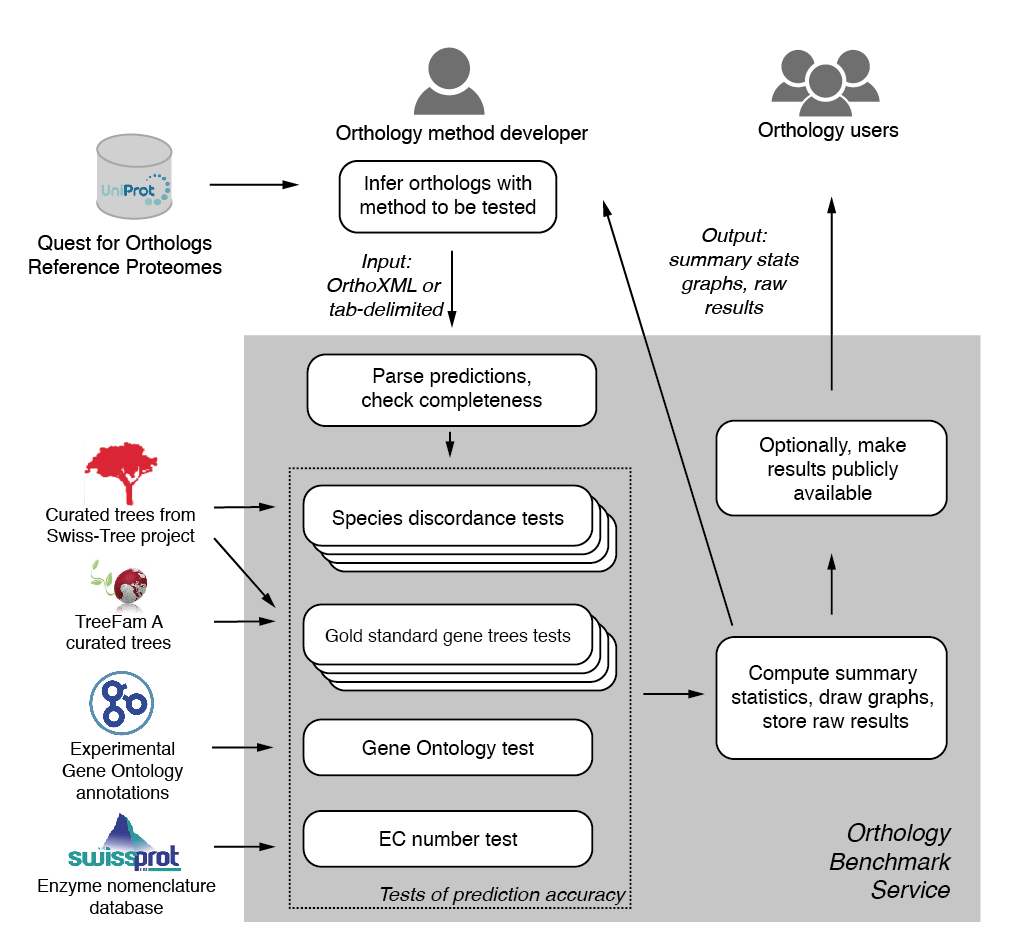
How does it work?
An orthology method developer should first infer the orthologs using the reference proteome dataset. The service will assess the induced pairwise orthologous relations. Therefore, the method developer must provide the predictions in a format from which the pairwise orthologous predictions can be extracted in an unambiguous way.
Once the predictions have been uploaded, the service ensures that only predictions among valid reference proteomes are provided. Benchmarks are then selected and run in parallel. Finally, statistical analyses of method accuracy are performed on each benchmark dataset. The raw data and summary results in form of precision-recall curves are stored and provided to the submitter.
The code and data that is used to run the benchmark is available on Github.
QfO Reference Proteomes Dataset
Orthology inference is most often based on molecular protein sequences. For a comparison of different orthology prediction methods, a common set of sequences must be established. Therefore, only identical proteins are mapped to each other.
To make comparisons of method easier, the orthology research community has agreed in 2009 to establish a common QfO reference proteome dataset. Currently, we are using the reference proteomes from 2020_04 and 2022_02 for benchmarking. The older datasets can no longer be used for benchmarking, but the results of the public projects stay for reference.
We encourage to use always the newest dataset: Established resources should run their pipeline on all datasets, including the cutting edge dataset 2022_02. Emerging tools and resources should at least use the latest stable 2020_04 dataset, which allows to compare the predictions against a large set of established orthology inference resources.
All releases of the QfO Reference Proteomes are available from UniProtKB's archive FTP server. The currently recommended datasets for benchmarking are:
Both datasets are recommended for benchmarking.Formats
Our benchmarks assess orthology on the bases of protein pairs. Therefore, we ask our users to upload their prediction in a format from which we can extract pairwise relations in an unambiguous manner: We support
- simple text file with two tab-separated columns of orthologous protein represented by their ids
- orthoxml v0.3 , which allows for nested orthologGroups and paralogGroups.
How to cite the orthology benchmark service
Adrian M Altenhoff, Brigitte Boeckmann, Salvador Capella-Gutierrez, Daniel A Dalquen, Todd DeLuca, Kristoffer Forslund, Jaime Huerta-Cepas, Benjamin Linard, Cécile Pereira, Leszek P Pryszcz, Fabian Schreiber, Alan Sousa da Silva, Damian Szklarczyk, Clément-Marie Train, Peer Bork, Odile Lecompte, Christian von Mering, Ioannis Xenarios, Kimmen Sjölander, Lars Juhl Jensen, Maria J Martin, Matthieu Muffato, Toni Gabaldón, Suzanna E Lewis, Paul D Thomas, Erik Sonnhammer, Christophe Dessimoz.
Standardized benchmarking in the quest for orthologs.
Nature Methods, 2016, 13, 425-430
![]() Full text
Full text
Adrian M Altenhoff, Javier Garrayo-Ventas, Salvatore Cosentino, David Emms, Natasha M Glover, Ana Hernández-Plaza, Yannis Nevers, Vicky Sundesha, Damian Szklarczyk, José M Fernández, Laia Codó, the Quest for Orthologs Consortium, Josep Ll Gelpi, Jaime Huerta-Cepas, Wataru Iwasaki, Steven Kelly, Odile Lecompte, Matthieu Muffato, Maria J Martin, Salvador Capella-Gutierrez, Paul D Thomas, Erik Sonnhammer, Christophe Dessimoz
The Quest for Orthologs benchmark service and consensus calls in 2020.
Nucleic Acids Res, 2020, 48:W1, W538–W545
![]() Full text
Full text
Yannis Nevers, Tamsin E M Jones, Dushyanth Jyothi, Bethan Yates, Meritxell Ferret, Laura Portell-Silva, Laia Codo, Salvatore Cosentino, Marina Marcet-Houben, Anna Vlasova, Laetitia Poidevin, Arnaud Kress, Mark Hickman, Emma Persson, Ivana Piližota, Cristina Guijarro-Clarke, the OpenEBench team the Quest for Orthologs Consortium , Wataru Iwasaki, Odile Lecompte, Erik Sonnhammer, David S Roos, Toni Gabaldón, David Thybert, Paul D Thomas, Yanhui Hu, David M Emms, Elspeth Bruford, Salvador Capella-Gutierrez, Maria J Martin, Christophe Dessimoz, Adrian Altenhoff
The Quest for Orthologs orthology benchmark service in 2022.
Nucleic Acids Res, 2022,
![]() Full text
Full text
Powered by OpenEBench
 All the computations are performed using the OpenEBench platform of ELIXIR.
All the computations are performed using the OpenEBench platform of ELIXIR.
OpenEBench is an infra-structure designed to establish a continuous automated benchmarking system for bioinformatics methods, tools and web services. It is being developed so as to cater for the needs of the bioinformatics community, especially software developers who need an objective and quantitative way to inform their decisions as well as the larger community of end-users, in their search for unbiased and up-to-date evaluation of bioinformatics methods.
Contact information
For questions, comments or to notify us about a problem, please contact us using the following form:
Funding of the orthology benchmarking service
![]() The benchmarking service receives funding from the Swiss Institute of Bioinformatics Service and Infrastructure grant as part of the SwissOrthology initiative.
The benchmarking service receives funding from the Swiss Institute of Bioinformatics Service and Infrastructure grant as part of the SwissOrthology initiative.